MYCOPLASMA Example of gene finding with MATLAB
This demonstration looks at some algorithms for finding ORFs and for assessing their significance.
Contents
Introduction
Mycoplasmas are members of the class Mollicutes and comprise a large group of bacteria which lack a cell wall, have small genomes and a characteristically low G+C content. Mycoplasmas are of interest because they are believed to represent a minimal life form, having yielded to selective pressure to reduce genome size. The species with the smallest genome size in this class is Mycoplasma genitalium (580 kb).
Reading files in FASTA FORMAT
The dna sequence of M. genitalium can be obtained from the EntrezSite with the accession number NC_000908. Using the fastaread function the FASTA format file can be read.
entrezSite = 'http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?'; textOptions = '&txt=on&view=fasta'; genbankID = '&list_uids=NC_000908'; mgen = fastaread([entrezSite textOptions genbankID]); % mgen.Header is the header information. % mgen.Sequence is the dna sequence stored as a string of characters.
If you don’t have a live web connection, you can load the data from a MAT-file using the command
% mgen=fastaread('Mgen_G37_.fasta'); % <== Uncomment this if no internet connection
This extracts the sequence.
seq=mgen.Sequence;
Exploring the Open Reading Frames (ORFs)
In a nucleotide sequence an obvious thing to look for is if there are any open reading frames. The function seqorfs can be used to determine the ORFs in a sequence.
orf=seqorfs(seq);
The variable orf is a structure with information about the start/stop positions of each ORFs, its length and which reading frame it is in. Here the minimum number of codons for an ORF to be considered valid is 10 (default value).
The minimum number of codons for an ORF to be considered valid can be also set. Here a threshold of 90 and 100 amino acids are considered leading to about 543 and 471 ORFs respectively. The original genome paper gave the number of genes as about 470.
[orf n_90]=seqorfs(seq,'MINIMUMLENGTH',90);
n_90
n_90 = 543
[orf n_100]=seqorfs(seq,'MINIMUMLENGTH',100);
n_100
n_100 = 471
You can look for the total number of ORFs
[orf n]=seqorfs(seq,'MINIMUMLENGTH',1);
n
n =
11922
Detecting significant ORFs
The classic approach to decide whether an ORF is a good candidate as a gene is to calculate the probability of seeing an ORF of a certain length L in a random sequence. To test the significance of ORFs a single-nucleotide permutation test can be used.
[orf1 n1]=seqorfs(seq(randperm(length(seq))),'MINIMUMLENGTH',1);
n1
n1 =
17324
The ORF length of the original and of the randomized sequences can be compared.
ORFLength=[]; for i=1:6 ORFLength=[ORFLength; orf(i).Length']; end ORFLength1=[]; for i=1:6 ORFLength1=[ORFLength1; orf1(i).Length']; end
[n,xout]=hist(ORFLength(ORFLength<450),30); [n1,xout1]=hist(ORFLength1(ORFLength1<450),30); bar(xout1,n1,0.5,'r') hold on bar(xout,n,0.5) hold off xlabel('ORF Length (in codons)'); title('ORFs Length distribution in M.Genitalium (blue) and randomised M.Genitalium (red)');
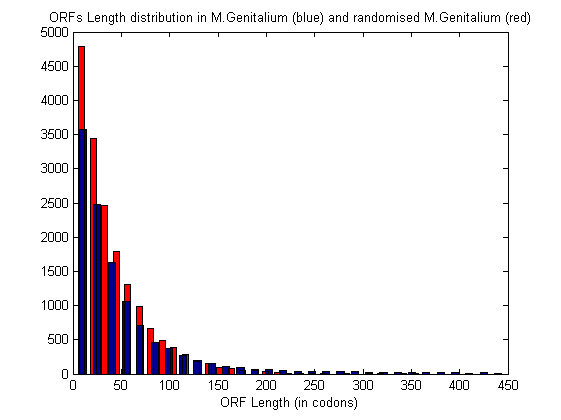
A threshold must be chosen in order to keep as candidate genes those ORFs in the M. Genitalium genome that are longer than most (or all) the ORFs in the random sequence.
max_threshold=max(ORFLength1) n_max=length(find(ORFLength>=max_threshold))
max_threshold = 360 n_max = 381
You can also use a more tolerant threshold in order to keep all ORFs of length equal to or greater than the top 5% of random ORFs.
empirical_threshold=prctile(ORFLength1,95) n_emp=length(find(ORFLength>=empirical_threshold))
empirical_threshold =
114
n_emp =
1520